BioGNN
Modèle de Deep Learning pour la prédiction d'activité biologique et de toxicité de molécules. Les interactions physico-chimiques complexes sont simulées par un réseau de neurones graphique, exploitant les dernières avancées de Deep Learning avec une architecture branchée et des mécanismes d'attention. Le modèle est déployé dans une interface utilisateur simple d'utilisation.
Le développement d’un nouveau médicament coûte en moyenne 2,2 milliards de dollars et prend plus d’une dizaine d’année, dont une large part est consacrée à tester des molécules qui s’avèrent inactives ou toxiques en phase tardive.
Et si on pouvait filtrer ces candidats bien plus tôt ?
C’est la question au cœur de BioGNN, qui utilise le Deep Learning pour prédire l’activité biologique d’une molécule directement depuis sa structure, avant même d’ouvrir un flacon en laboratoire.
Les domaines d’application sont larges : industrie pharmaceutique, biostimulation agricole, toxicologie, cosmétique.
Approche technique
Données
Les données proviennent de ChemBL, la plus grande base publique de résultats d’essais biologiques. Chaque entrée associe une molécule (structure SMILES) à un résultat d’activité mesuré in vitro sur une cible biologique spécifique.
Le dataset a été filtré, équilibré et préparé via RDKit pour extraire les caractéristiques atomiques et les matrices d’adjacence nécessaires à l’entraînement.
Un choix a été fait de se limiter à des groupes d’organismes modèles couramment utilisés en biologie pour maximiser la pertinence phylogénique des prédictions.
Représentation moléculaire par graphes
Une molécule est naturellement un graphe : les atomes sont les nœuds, les liaisons chimiques sont les arêtes. Cette représentation capture l’information structurelle complète, et bien mieux que les fingerprints ECFP classiques ou les représentations SMILES linéaires.
BioGNN exploite directement cette structure via des Graph Neural Networks (GNN), qui propagent l’information le long des liaisons pour construire une représentation globale de la molécule.
Architecture branchée avec mécanismes d’attention
L’architecture s’inspire des avancées en IA générative :
- Couches de message-passing : agrégation des caractéristiques des atomes voisins à chaque étape
- Mécanismes d’attention : pondération des contributions de chaque atome selon leur pertinence biologique
- Architecture branchée : plusieurs branches parallèles spécialisées, fusionnées avant les têtes de décision
- Têtes de décision multiples : classification binaire (actif / inactif) des quatre propriétés simultanément
Résultats
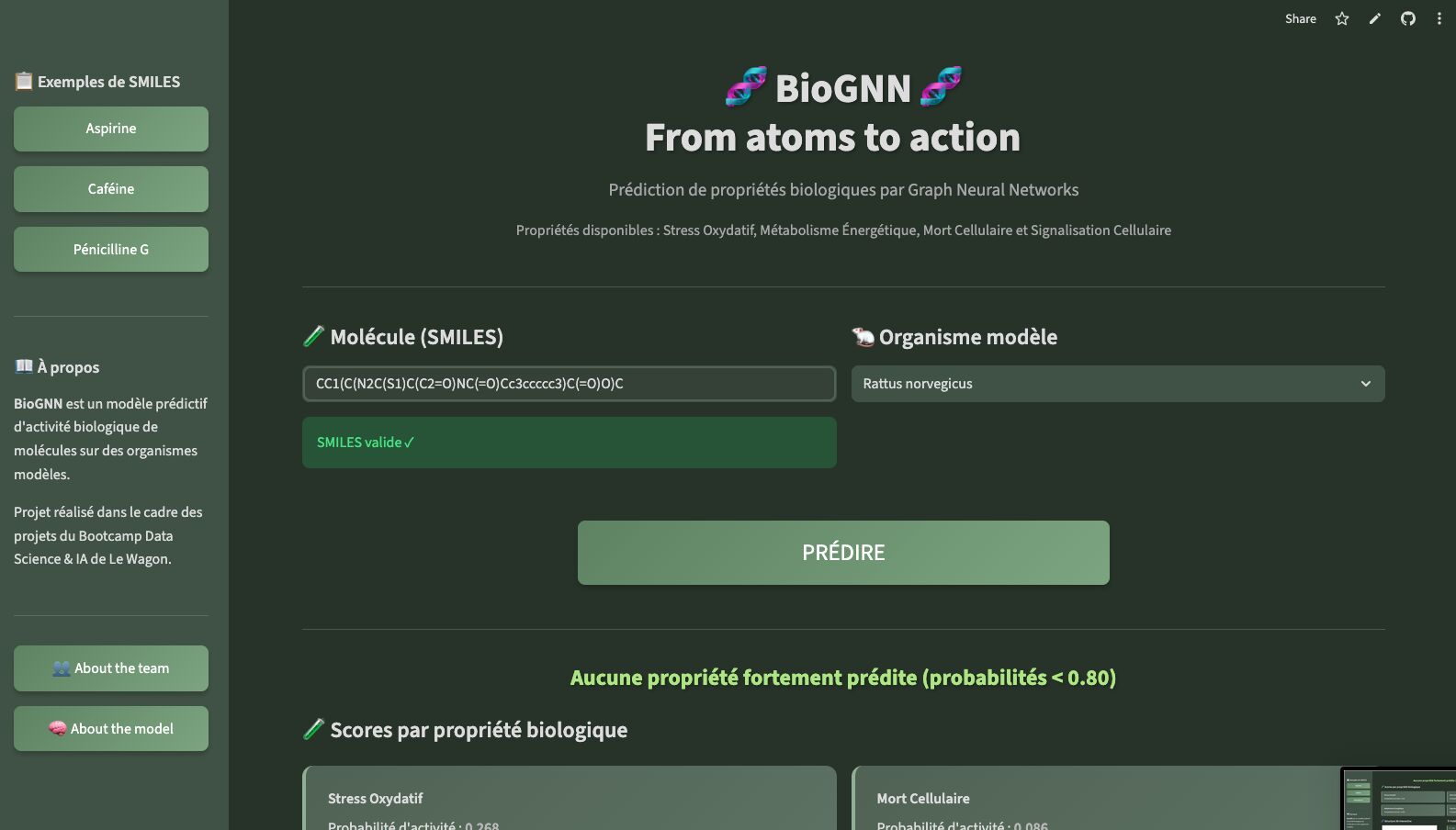

Le projet a abouti en 10 jours à deux modèles complémentaires et une interface utilisateur fonctionnelle permettant de soumettre n’importe quelle molécule en format SMILES et d’obtenir une prédiction d’activité avec score de confiance, et dans le cas de molécules toxiques pour l’organisme étudié, un message d’alerte.
Équipe et Stack technique
Projet réalisé dans le cadre du bootcamp Data Science & IA du Wagon. La présentation finale est disponible sur YouTube.
Projet réalisé avec :
| Outil | Rôle |
|---|---|
| RDKit | Parsing SMILES, génération de graphes moléculaires, calcul de descripteurs |
| PyTorch | Construction et entraînement du modèle |
| PyTorch Geometric | Couches GNN spécialisées |
| Optuna | Optimisation des hyperparamètres |
| Streamlit | Dashboarding |
Ressources
- Predicting bioactivity — Cambridge MedChem Consulting
- Estimated R&D cost per drug (2009-2018)
- R&D cost per drug in 2024 — Deloitte
- Hands-On Graph Neural Networks Using Python — Packt
- Node Classification with GNNs in Keras